(Prof. Dr. V. Hach-Wunderle)
Hämostaseologisches Risikoprofil bei Venenthrombose
Viola Hach-Wunderle
Das Auftreten einer Venenthrombose kann auf eine thrombophile Diathese hinweisen. Das gilt insbesondere für die Manifestation im jungen Lebensalter, für rezidivierende Thrombosen, bei einer familiären Disposition und bei außergewöhnlicher Lokalisation der Krankheit. Die Basis-Diagnostik besteht in der Bestimmung der Gerinnungsinhibitoren, der APC-Ratio, der An-tiphospholipid-Antikörper sowie der molekulargenetischen Untersuchung von Prothrombin. Das Thromboserisiko wird bei der homozygoten Faktor V Leiden-Mutation, beim angeborenen Antithrombin-Mangel und bei bestimmten Multigen-Defekten als besonders hoch eingestuft.
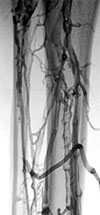
Phlebothrombose vom aszendierenden Verlaufstyp. Darstellung durch die aszendierende Phlebographie mit digitaler Technik. Indikation zur differenzierten hämostaseologischen Abklärung und zur antikoagulatorischen Therapie.
Die tiefe Venenthrombose (TVT) gehört trotz aller prophylaktischer Maßnahmen weiterhin zu den häufigsten Krankheiten unserer Zeit. Die jährliche Inzidenz wird mit 1-2 pro 1000 in der Normalbevölkerung angegeben.
Eine strukturierte Anamnese trägt maßgeblich zur Einschätzung des individuellen Thromboserisikos bei (73). Dank der modernen bildgebenden Untersuchungsmethoden lässt sich die Diagnose heute frühzeitig und zuverlässig stellen. Spezielle hämostaseologische Tests decken in vielen Fällen angeborene oder erworbene Defekte auf, die für das Auftreten von Rezidivthrombosen von großer Bedeutung sind. Vor diesem Hintergrund gewinnt eine individuell angepasste medikamentöse Thromboseprophylaxe einen neuen Stellenwert.
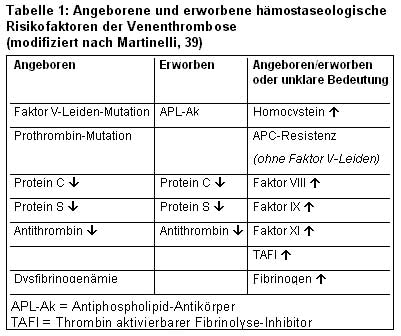
Die Venenthrombose ist ein komplexes Krankheitsbild. Eine Vielzahl von angeborenen und erworbenen Risikofaktoren kann zu ihrer Entstehung beitragen (20-22). Dabei ist auch heute noch die sogenannte Virchow`sche Trias mit den Komponenten Gefäßwandschaden, verlangsamter Blutströmung und veränderter Zusammensetzung des Blutes für unsere Vorstellung von der Thrombogenese von Bedeutung; aber die molekularen Abläufe sind im einzelnen noch nicht ausreichend erforscht (Tabelle 1).
Angeborene Thrombophilie
Den angeborenen thrombophilen Gerinnungsstörungen kommt heute eine große klinische Bedeutung zu. Dazu zählen die Verminderung von Antithrombin, Protein C und Protein S sowie die Faktor V-Leiden-Mutation und die Prothrombin-Mutation.
Verminderung von Antithrombin,Protein C und
Protein S
In den Jahren 1965 bis 1984 wurde der Zusammenhang zwischen einem angeborenen Mangel von Antithrombin (13,15,64) bzw. von Protein C oder Protein S (3,19,57) und einer Neigung zur Venenthrombose erkannt.
Durch funktionelle und immunologische Untersuchungen lassen sich bei den drei Defekten spezielle Typen differenzieren, die mit einem unterschiedlichen Thromboserisiko einhergehen. Beim Typ I beruht der Mangel jeweils auf einer verminderten Synthese in der Leber (=quantitativer Defekt). Beim Typ II ist zwar die Konzentration des Eiweißkörpers im Plasma normal, die biologische Aktivität jedoch wegen einer abnormen Molekülstruktur vermindert (=qualitativer Defekt). Beim Protein S-Mangel wird zusätzlich berücksichtigt, dass Protein S einerseits in freier Form und andererseits in einem Komplex mit C4-Bindungsprotein zirkuliert. Das führt dann zur Differenzierung von drei Typen, wobei die Protein S-Aktivität bei allen Formen vermindert ist. Die gesamte und die freie Konzentration von Protein S sind beim Typ I vermindert und beim Typ II normal; beim Typ III ist die freie Konzentration erniedrigt.
Der homozygote Mangel an Protein C oder Protein S kann eine ausgeprägte Thromboseneigung verursachen, wie beispielsweise die neonatale Purpura fulminans oder die Kumarin-induzierte Hautnekrose (45,58). Der heterozygote Mangel eines Gerinnungsinhibitors führt oft zur Erstmanifestation einer Venenthrombose im Lebensalter unter 40 Jahre. Dabei sind gelegentlich atypische Gefäßregionen betroffen wie zerebrale Sinusvenen, abdominelle Venen und Armvenen. Die Betroffenen neigen zu Rezidivthrombosen und haben häufig eine positive Familienanamnese (11).

Von den drei Gerinnungsinhibitoren ist das Thromboserisiko beim Antithrombin-Mangel relativ am höchsten einzustufen (35), gefolgt vom Protein C-Mangel.
Faktor V-Leiden-Mutation
Die Resistenz von aktiviertem Faktor V gegenüber seiner proteolytischen Spaltung durch aktiviertes Protein C (APC) wurde erstmals 1993 beschrieben (10). In ca. 95% der Fälle ist dafür eine Punktmutation mit Austausch einer Aminosäure (Glutamin statt Arginin) in Position 506 von Faktor V (F.V: Q506) verantwortlich. Nach dem Entdeckungsort in den Niederlanden wird diese Mutation als Faktor V-Leiden bezeichnet (2).
Die Faktor V-Leiden-Mutation ist die häufigste Ursache einer Thrombophilie. Die Prävalenz des Defekts ist in der kaukasischen Bevölkerung relativ hoch; sie beträgt 2 bis 15% in der Gesamtbevölkerung und bis zu 45% in selektierten Patientenkollektiven (18,31,62). Der Vererbungsmodus ist autosomal dominant. Das Risiko einer Erstmanifestation der Venenthrombose ist bei heterozygoten Merkmalsträgern bis zu 7-fach und bei homozygoter Ausprägung bis zu 80-fach erhöht im Vergleich zu Gesunden (54). Patienten mit Faktor V-Leiden-Mutation haben oft eine relativ milde Thromboseneigung; es können oberflächliche Venenthrombosen auftreten (42,53) und Lungenembolien sind relativ selten (41,66,70). Die Erstmanifestation einer Thrombose findet häufig erst in fortgeschrittenem Alter statt (43).
In letzter Zeit wurden andere genetische Defekte am Faktor-V-Molekül entdeckt, die ebenfalls eine APC-Resistenz verursachen, z.B. Faktor V-Cambridge und Faktor V-Hongkong (7,74).
Prothrombin-Mutation G20210A
Eine Mutation an der Nukleotid-Position 20210 der 3`-Region des Prothrombin-Gens wurde 1996 entdeckt (50). Bei den Merkmalsträgern ist der Prothrombin-Plasmaspiegel um ca. 30% erhöht (50); daraus resultiert ein erhöhtes Potential für die Thrombinbildung (33). Aber auch erhöhte Prothrombin-Spiegel ohne Nachweis einer Mutation scheinen mit einem erhöhten Thromboserisiko einherzugehen (60).
Die Mutation kommt bei 2 bis 4% der kaukasischen Bevölkerung vor. Die Prävalenz ist in Südeuropa etwa doppelt so hoch wie in Nordeuropa (53). Dieser geographische Gradient ist genau umgekehrt zur Faktor-V-Leiden-Mutation.
Bei selektierten Patienten mit TVT wird eine Prävalenz der Mutation bis zu 20% mitgeteilt; das relative Thromboserisiko ist bei Merkmalsträgern 2 bis 4-fach höher als bei Kontrollpersonen (4,8,23,37,38,50).
Multigen-Defekte
Bei der relativ hohen Frequenz von Faktor V-Leiden-Mutation und Prothrombin-Mutation in der kaukasischen Bevölkerung sind kombinierte Defekte keineswegs selten. Die Betroffenen haben von vornherein ein höheres Thromboserisiko (40). Bei der Kombination beider Defekte ist das relative Risiko für Rezidivthrombosen um das 2,6-fache erhöht im Vergleich zu Patienten mit alleiniger Faktor V-Leiden-Mutation oder fehlendem Nachweis eines Defekts. Bei gleichzeitigem Auftreten einer spontanen Thrombose ist das Risiko noch einmal deutlich höher (12). Auch andere Kombinationen von Gerinnungsdefekten wurden beschrieben, am häufigsten in Assoziation mit der Faktor V-Leiden-Mutation.
Dysfibrinogenämie
Die kongenitale Dysfibrinogenämie ist durch die Biosynthese eines strukturell abnormen Fibrinogenmoleküls charakterisiert. Bisher sind etwa 300 Fälle in der Weltliteratur beschrieben (14,47). In ca. 55% der Familien liegt keine klinische Symptomatik vor, 25% weisen eine Blutungsneigung und 20% eine Thromboseneigung auf.
Erworbene Thrombophilie
Die größte Bedeutung kommt den Antiphospholipid-Antikörpern zu. Bei allen anderen Defekten ist der Stellenwert in Bezug auf ein erhöhtes Thromboserisiko weniger gut belegt.
Antiphospholipid-Antikörper
Antiphospholipid-Antikörper (APL-Ak) sind eine heterozygote Gruppe von Antikörpern, die sich mit Hilfe von Phospholipid-bindenden Plasmaproteinen wie Beta2-Glykoprotein I an Oberflächen anlagern. Es werden Lupus-Antikoagulanzien und Antikardiolipin-Antikörper differenziert. Eine Erhöhung ist gelegentlich beim systemischen Lupus erythematodes oder bei anderen Autoimmunkrankheiten vorhanden; oftmals kann aber keine Grundkrankheit nachgewiesen werden. Die Antikörper werden in der Regel erworben; in Einzelfällen wurde über eine familäre Vererbung berichtet.
Die Prävalenz in der Normalbevölkerung ist nicht bekannt. Bei Patienten mit TVT wird sie mit 5 bis 15% angegeben (17,44,59).
Charakteristischerweise kommen bei Patienten mit APL-AK sowohl venöse als auch arterielle Thrombosen vor. Auch rezidivierende Aborte sind typisch. Das Thromboserisiko ist bei Vorlie-gen von Antikardiolipin-Antikörpern auf das 2-fache und bei Lupus-Antikoagulanzien auf das 5 bis 10-fache erhöht (71,72). In prospektiven Studien wurde ein 2-fach erhöhtes Risiko für rezidivierende Thrombosen nach Beendigung der oralen Antikoagulation beobachtet (27,56).
APC-Resistenz (ohne Faktor V-Leiden-Mutation)
Zu den erworbenen Ursachen einer APC-Resistenz zählen beispielsweise die Schwangerschaft und die Einnahme von oralen Antikonzeptiva (9,55), aber auch Adipositas, Hypercholesterinämie und Hyperfibrinogenemie (65). In einer bevölkerungsbasierten Studie an mehr als 15000 Personen konnte gezeigt werden, dass das relative Thromboserisiko bei erworbener APC-Resistenz doppelt so hoch ist im Vergleich zu Personen ohne Gerinnungsdefekt (52). In einem unselektierten Patientenkollektiv ist damit zu rechnen, dass 1 von 10 Patienten mit TVT den Defekt aufweist. Deshalb sollte die sogenanne APC-Ratio als funkioneller Screening-Test beibehalten und der molekulargenetischen Untersuchung von Faktor V vorangestellt werden.
Hyperhomocysteinämie
Homocystein wird via Methylen-Tetrahydrofolat-Reduktase (MTHFR) Vitamin B12-abhängig in Methionin bzw. via Cystation-Beta-Synthase (CBS) Vitamin B6-abhängig in Cystein überführt. Erhöhte Homocysteinspiegel finden sich somit hereditär bei einem Defekt der MTHFR oder der CBS. Erworbene Defekte treten bei einem Mangel an Vitamin B12, B6 oder Folsäure sowie u.a. auch bei Niereninsuffizienz, Schwangerschaft und bei Malignomen auf. Ein erhöhter Homocysteinspiegel lässt sich durch Substitution von Folsäure, Vitamin B12 oder Vitamin B6 senken. Es ist aber unklar, ob damit auch das Thromboserisiko abnimmt.
Der mögliche Zusammenhang zwischen einer milden Hyperhomocysteinämie und einer Neigung zu Venenthrombosen wurde erstmals 1994 beschrieben; es können arterielle und venöse Thrombosen auftreten (5). In Fall-Kontroll-Studien ergab sich ein 2,5-fach erhöhtes Risiko für die Erstmanifestation einer Thrombose bei milder Ausprägung des Defekts (5). In einer prospektiven Studie fand sich ein 2.7-fach erhöhtes Risiko für Rezidivthrombosen nach Absetzen der gerinnungshemmenden Behandlung (16). Während zwei Arbeitsgruppen eine Thromboseneigung bei der Kombination einer Hyperhomocysteinämie oder einer homozygoten MTHFR-Mutation mit einer Faktor V-Leiden-Mutation beobachteten (6,51), bestätigte sich das nicht in einer anderen Untersuchung (28). Die genaue Bedeutung für das Auftreten von venösen Thrombosen ist also noch unklar.
Erhöhung von Faktor VIII
Erhöhte Plasmaspiegel von Faktor VIII gelten neuerdings als Risikofaktor für die Manifestation von venösen Thrombosen (29,32,34,48,49). Ein ursächlicher Zusammenhang wurde postuliert, nachdem sich zeigte, dass zwischen Faktor VIII einerseits und sogenannten Akute-Phase-Proteinen wie C-reaktives Protein und Fibrinogen andererseits keine unmittelbare Relation vorlag (48,49).
Die Prävalenz von hohen Faktor VIII-Werten bei Patienten mit Thrombosen variiert zwischen 19 und 25% (29,32). Dabei wurde als sogenannter cut off-Wert die 90% Perzentile der Messwerte in einem Normalkollektiv zugrunde gelegt, entsprechend einem Faktor VIII-Spiegel >150 IU/dl (29) oder 175 IU/dl (32). Die erhöhten Faktor VIII-Plasmaspiegel können per-sistieren. Bei deutlich erhöhten Messwerten >230 IU/dl wurde ein erhebliches Risiko für Rezi-divthrombosen nach Absetzen der oralen Antikoagulation nachgewiesen (32,24). Möglicherweise ergeben sich daraus Schlussfolgerungen bezüglich der optimalen Dauer einer gerinnungshemmenden Behandlung. Prospektive Untersuchungen zu dieser Thematik stehen derzeit noch aus.
Erhöhung von Faktor IX bzw. Faktor XI
Das Thromboserisiko ist bei erhöhten Plasmaspiegeln von Faktor IX und Faktor XI verdoppelt (46,68). Die Prävalenz liegt bei 20% bzw. bei 19% im Vergleich zu Personen mit Messwerten unterhalb der 90%-Perzentile eines Normalkollektivs. Eine familiäre Disposition wurde bisher nicht nachgewiesen.
Erhöhung des durch Thrombin aktivierbaren Fibrinolyseinhibitors (TAFI)
Die Prävalenz einer erhöhten Konzentration des durch Thrombin aktivierbaren Fibrinolyseinhibitors (TAFI) bei Patienten mit Thrombosen wird auf 14% geschätzt; das Thromboserisiko ist offenbar nur leicht erhöht (69).
Erhöhung von Fibrinogen
Eine Erhöhung von Fibrinogen (über 5 g/l) soll mit einem 4-fach erhöhten Thromboserisiko einhergehen und zwar unabhängig von anderen Risikofaktoren wie erhöhtem Lebensalter und Tumorkrankheit (30).
Erworbener Mangel an Antithrombin, Protein C oder Protein S
Bei der Verminderung eines oder mehrerer Gerinnungsinhibitoren wird eine erhöhte Disposition zu Thrombosen angenommen, die in Abhängigkeit von der zugrunde liegenden Situation stark variiert (26,67). So ist bei schweren Leberkrankheiten aufgrund gleichzeitiger Defekte der plasmatischen und der thrombozytären Blutgerinnung kein erhöhtes Thromboserisiko zu erwarten. Anders verhält es sich aber beispielsweise bei der disseminierten intravasalen Gerinnung; dabei sind Thrombosen in den kleinen Gefäßen geradezu charakteristisch.
Eine relevante Verminderung von Antithrombin ist beim nephrotischen Syndrom sowie bei der Therapie mit Östrogenen bzw. mit L-Asparaginase zu erwarten. Protein C und Protein S sind beim Vitamin K-Mangel vermindert und Protein S darüber hinaus in der Schwangerschaft und bei Einnahme von oralen Antikonzeptiva (26,63).
Praktische Vorgehensweise bei der Thrombophilie-Diagnostik
Nachfolgend sollen einige Fragestellungen besprochen werden, die von praktischer Relevanz sind.
Zeitpunkt der Untersuchung
Das Thrombophilie-Screening kann am Tag der Thrombose-Diagnostik erfolgen. Normalbefunde schließen dann eine thrombophile Diathese nach dem aktuellen Kenntnisstand aus. Eine Verminderung von Antithrombin, Protein C oder Protein S bzw. eine Erhöhung von einzelnen Gerinnungsfaktoren wie Faktor VIII oder Fibrinogen bedarf der späteren Kontrolle, da eine Beeinflussung durch die akute Krankheitsphase nicht auszuschließen ist. Molekulargenetische Untersuchungen von Faktor V und von Prothrombin sind davon immer unbeeinflusst, auch die Bestimmung von Antiphospholipid-Antikörpern. Unter Therapie mit Heparin kann Antithrombin vermindert sein. Bei Behandlung mit oralen Antikoagulanzien sind Protein C und Protein S nicht sicher zu beurteilen. Die spezielle Diagnostik muß dann nach Absetzen der gerinnungshemmenden Behandlung bei normaler INR (International Normalized Ratio) und normaler APTT (aktivierter partieller Thromboplastinzeit) vorgenommen werden. Wenn das Risiko einer Rezidivthrombose zu diesem Zeitpunkt als hoch angesehen wird, kann die Diagnostik dann auch unter prophylaktischem Heparinschutz erfolgen. In der Schwangerschaft und während der Einnahme von oralen Antikonzeptiva sind die Protein S-Spiegel vermindert.
Untersuchungsprogramm
Das Untersuchungsprogramm beinhaltet die globalen Tests APTT, Thromboplastinzeit und Thrombozytenzahl. Die Gerinnungsinhibitoren Antithrombin, Protein C und Protein S werden zunächst funktionell untersucht; erst bei wiederholt pathologischem Befund schließt sich ein immunologischer Test an. Die APC-Ratio stellt die Basis-Diagnostik einer APC-Resistenz dar. Bei einer Verminderung erfolgt die Molekulargenetik von Faktor V. Der Ausschluß einer Fak-tor V Leiden-Mutation bei pathologischer APC-Resistenz spricht für einen anderen hereditären Defekt (z:B. Faktor V Cambridge) oder für eine erworbene Störung. Der Nachweis oder Ausschluß der Prothrombin-Mutation G20210A erfolgt von vornherein molekulargenetisch. Die Antiphospholipid-Antikörper (APL-AK) beinhalten die Lupus-Antikoagulanzien und die Anticardiolipin-Antikörper; die Lupus-Antikoagulanzien sollten wegen der unzureichenden Treffsicherheit eines einzelnen Tests mit mindestens zwei Verfahren untersucht werden. Eine Dysfibrinogenämie ist selten; ein erster Hinweis ergibt sich durch ein vermindertes Fibrinogen nach CLAUSS. Die Höhe des Faktor VIII-Spiegels ist zukünftig vielleicht für die Dauer einer gerinnungshemmenden Behandlung mit ausschlaggebend; größere prospektive Studien stehen derzeit noch aus. Andere thrombophile Faktoren sind in Bezug auf ein erhöhtes Thromboserisiko noch nicht ausreichend evaluiert.
Untersuchungskollektiv
Die Thrombophilie-Diagnostik sollte vor allem bei jüngeren Patienten erfolgen; meist wird ein Lebensalter unter 45 Jahren bei Erstmanifestation der Thrombose genannt. Auch eine auffälligen Familienanamnese bezüglich thromboembolischer Krankheiten ist relevant und zwar insbesondere dann, wenn eine zusätzliche Risikosituation hinzukommt wie geplante Einnahme von oralen Antikonzeptiva oder Schwangerschaft. Abklärungsbedürftig erscheint auch die Manifestation einer Thrombose ohne andere disponierende Ursachen sowie der rezidivierende Krankheitsverlauf. Eine Thromboselokalisation in einer ungewöhnlichen Gefäßregion weist ebenfalls auf eine thrombophile Diathese hin. Die Kumarinnekrose kann auf einem Protein C- oder Protein S-Mangel, seltener auf einem Antithrombin-Mangel beruhen. Das Auftreten von arteriellen und venösen Thrombosen bei einem Patienten ist typisch für APL-AK, auch für die seltene Dysfibrinogenämie und die Hyperhomozysteinämie. Rezidivierende Aborte können beim Antiphospholipid-Antikörper-Syndrom auftreten.
Bewertung der Untersuchungsergebnisse
Das relativ höchste Thrombose-Risiko wird für den angeborenen Antithrombin-Mangel, die homozygote APC-Resistenz und bestimmte Multigen-Defekte angenommen (Tabelle 3). In diesen Fällen ist eine sorgfältige und ausreichend hoch dosierte Thrombose-Prophylaxe in Risikosituationen angezeigt; nach einer ersten Thrombose ist die Langzeit-Antikoagulation in Erwägung zu ziehen. Das gilt auch für die APL-AK, die ausgesprochen thrombogen sein können. Bei allen anderen Defekten wird die gerinnungshemmende Therapie nach der ersten Thrombose wieder beendet; es erfolgt dann eine medikamentöse Prophylaxe in Risikosituationen. Noch unklar ist die Vorgehensweise bei persistierend erhöhten Faktor VIII-Werten in der Folge einer Thrombose.

Literaturverzeichnis
1. Bertina RM (2001) Genetic approach to thrombophilia. Thromb Haemost 86: 92-103
2. Bertina RM, Koeleman RPC, Koster T Rosendaal FR, et al (1994) Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 369: 64-74
3. Broekmans AW, Veltkamp JJ, Bertina RM (1983) Congenital protein C deficiency and venous thromboembolism: a study of three Dutch families. N Engl J Med 309: 340-344
4. Brown K, Luddington R, Williamson D, Baker P (1997) Risk of venous thromboembo-lism associated with a G to A transition at position 20210 in 3´untranslated region of the prothrombin gene. Br J Haematol 98: 907-909
5. Cattaneo M (1999) Hyperhomocysteinaemia, atherosclerosis and thrombosis. Thromb Haemost 81: 165-176
6. Cattaneo M, Monzani ML, Martinelli, Falcon CR, et al (1998) Interrelation of hyperhomocysteinaemia, factor V Leiden, and the risk of future venous thromboembolism. Circulation 97: 295-296
7. Chan WP, Lee CK, Kwong YL, Liang R (1998) A novel mutation of ARG306 of factor V-gene in Hong Kong Chinese. Blood 91: 1135-1139
8. Cumming AM, Keeney S, Salden A, Bhavnani M, et al (1997) The prothrombin gene G20210A variant: prevalence in a U.K. anticoagulant clinic population. Br J Haematol 98:353-355
9. Cumming AM, Tait RC, Fildes S, Yoong A, et al (1995) Development of resistance to activated protein C during pregnancy. Br J Haematol 90: 725-727
10. Dahlbäck B, Carlsson M, Svensson PJ (1993) Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 90: 1004-1008
11. De Stefano V, Finazzi G, Mannucci PM (1996) Inherited thrombophilia: pathogenesis, clinical syndromes, and management. Blood 87: 3531-3544
12. De Stefano V, Martinelli I, Mannucci PM, Paciaroni K, et al (1999) The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med 341: 801-806
13. Demers C, Ginsberg JS, Hirsh J, Henderson P, et al, (1992) Thrombosis in antithrom-bin III-deficient persons: report of a large kindred and literature review. Ann Intern Med 116: 754-761
14. Ebert R (1994) Index of variant human fibrinogens. Boca Raton, FL: CRC Press
15. Egeberg O (1965) Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 13: 516-530
16. Eichinger S, Stümpflen A, Hirschl M, et al (1998) Hyperhomocysteinemia is a risk factor of recurrent venous thromboembolism. Thromb Haemost 80: 566-569
17. Ginsberg JS, Wells PS, Brill-Edwards P, Donovan D, et al (1995) Antiphospholipid antibodies and venous thromboembolism. Blood 86: 3685-3691
18. Griffin JH, Evatt B, Wideman C, Fernandez JA (1993) Anticoagulant protein C path-way defective in a majority of thrombophilic patients. Blood 82: 1989-1993
19. Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, et al, (1981) Deficiency of protein C in congenital thrombotic disease 68: 1370-1373
20. Hach-Wunderle V (1995) Die Vielfalt der thrombophilen Risikofaktoren. Krankenhaus Arzt 68: 556-562
21. Hach-Wunderle V (1999) Prädisposition und Auslöser der venösen Thrombose. Wien Med Wschr 149: 35-36
22. Hach-Wunderle V, Scharrer I (1993) Prävalenz des hereditären Mangels an Anti-thrombin III, Protein C und Protein S. Dtsch Med Wschr 118: 187-190
23. Hillarp A, Zöller B, Svensson PJ, Dahlbäck B (1997) The 20210A allele of the pro-thrombin gene is a common risk factor among Swedish outpatients with verified deep venous thrombosis. Thromb Haemost 78: 990-992
24. Hirsh J, Martin HP, Samama M (1994) Approach to the thrombophilic patient. In: Colman RW, Hirsh J, Marder VJ, Salzman EW (eds) Haemostasis and Thrombosis: Basic principles and clinical practice. Lippincott, Philadelphia, S. 1543-1561
25. Kearon C (2001) Epidemiology of venous thromboembolism. Sem Vasc Med 1: 7-25
26. Kearon C, Crowther M, Hirsh J (2000) Management of patients with hereditary hypercoagulable disorders. Annu Rev Med 51: 169-185
27. Kearon C, Gent M, Hirsh J, et al (1999) Comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 340: 901-907
28. Kluijtmans LAJ, Den Heijer M, Reitsma PH, Heil SG, et al (1998) Thermolabile methylene-tetrahydrofolate reductase and factor V Leiden in the risk of deep-vein thrombosis. Thromb Haemost 79: 254-258
29. Koster T, Blann AD, Briet E, Vandenbroucke JP, et al (1995) Role of clotting factor VIII in effect of von Willebrand factor and occurrence of deep-vein thrombosis. Lancet 345: 152-155
30. Koster T, Rosendaal FR, Reitsma PH, van der Velden PA, et al (1994) Factor VII and fibrinogen levels as risk factors for venous thrombosis. Thromb Haemost 71: 719-722
31. Koster T, Rosendahl FR, de Ronde H, Briet E, et al (1993) Venous thrombosis due to a poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet 342: 1503-1506
32. Kraaijenhagen RA, in´t Anker PS, Koopman MM, Reitsma PH, et al (2000) High plasma concentration of factor VIIIc is a major risk factor for venous thromboembo-lism. Thromb Haemost 83: 5-9
33. Kyrle PA, Mannhalter C, Beguin S, Stümpflen A, et al (1998) Clinical studies and thrombin generation. Arterioscler Thromb Vasc Biol 18:1287-1291
34. Kyrle PA, Minar E, Hirschl M, Bialonczyk C, et al (2000) High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med 343: 457-462
35. Lane AD, Mannucci PM, Bauer AK, Bertina RM et al.(1996) Inherited thrombophilia: part 1. Thromb Haemost 76: 651-662
36. Lensing AWA, Prandoni P, Prins MH, Büller HR (1999) Deep-vein thrombosis. Lan-cet 353: 479-485
37. Leroyer C, Mercier B, Oger E, Chenu E, et al (1998) Prevalence of the 20210A allele of the prothrombin gene in venous thromboembolism patients.Thromb Haemost 80: 49-51
38. Margaglione M, Brancaccio V, Giuliani N, D`Andrea G, et al (1998) Increased risk for venous thrombosis in carriers of the prothrombin G20210A gene variant. Ann Intern Med 129: 89-93
39. Martinelli I (2001) Risk factors in venous thromboembolism. Thromb Haemost 86: 395-403
40. Martinelli I, Bucciarelli P, Margaglione M, De Stefano V, et al (2001) The risk of venous thromboembolism in family members with mutations in the genes of factor V, prothrombin or both. Br J Haematol, in press
41. Martinelli I, Cattaneo M, Panzeri D, Taioli E, et al (1997) Low prevalence of factor V: Q506 in 41 patients with isolated pulmonary embolism. Thromb Haemost 77: 440-3
42. Martinelli I, Cattaneo M, Taioli E, De Stefano V (1999) Genetic risk factors for super-ficial vein thrombosis. Thromb Haemost 82: 1215-1217
43. Martinelli I, Mannucci PM, De Stefano V, Taioli E, et al (1998) Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood 92: 2353-2358
44. Mateo J, Oliver A, Borrell M, Sala N, et al (1997) Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism - results of the Spanisch multicentric study on thrombophilia (EMET-study). Thromb Haemost 77: 444-451
45. Mc Gehee WG, Klotz TA, Epstein DJ, Rapaport SI (1984) Coumarin necrosis associated with hereditary protein C deficiency. Ann Intern Med 101: 59-60
46. Meijers CMJ, Tekelenburg LHW, Bouma NB, Bertina RM (2000) High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med 342: 696-701
47. Mosesson MW (1999) Dysfibrinogenemia and thrombosis. Sem Thromb Haemost 25: 311-319
48. O`Donnell J, Mumford AD, Manning RA, Laffan M (2000) Elevation of factor VIII:C in venous thromboembolism is persistent and independent of the acute phase response. Thromb Haemost 83: 10-13
49. O`Donnell J, Tuddenham EG, Manning R, Kemball-Cook G, et al (1997) High prevalence of elevated factor VIII levels in patients referred for thrombophilia screening: role of increased synthesis and relationship to acute phase reaction. Thromb Haemost 77: 825-828
50. Poort SR, Rosendaal FR, Reitsma PH, Bertina RM (1996) A common genetic varia-tion in the 3`-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 88: 3698-3703
51. Ridker PM, Hennekens CH, Selhub J, Miletich JP, et al (1997) Interrelation of hyperhomocysteinaemia, factor V Leiden, and the risk of future venous thromboembolism. Circulation 95: 1777-1782
52. Rodeghiero F, Tosetto A (1999) Activated protein resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Ann Intern Med 130: 643-650
53. Rosendaal FR, Doggen CJM, Zivelin A, Arruda VR, et al (1998) Geographic distri-bution of the 20210G to A prothrombin variant. Thromb Haemost 79: 706-708
54. Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH (1995) High risk of throm-bosis in patients homozygous for factor V Leiden. Blood 85: 1504-1508
55. Rosing J, Middeldorp S, Curvers J, Thomassen M, et al (1999) Low-dose oral contraceptives and acquired resistance to activated protein C: a randomised cross-over study. Lancet 354: 2036-2040
56. Schulman S, Svenungsson E, Granqvist S (1998) Anticardiolipin-antibodies predict early recurrence of thromboembolism and death among patients with venous thrombo-embolism following anticoagulant therapy. Am J Med 104: 332-338
57. Schwarz HP, Fischer M, Hopmeier P, Batard MA, et al, (1984) Plasma protein S deficiency in familial thrombotic disease. 64: 1297-1300
58. Seligsohn U, Berger A, Abend M, Rubin L, et al, (1984) Homozygous protein C deficiency manifested by massive venous thrombosis in the newborn. N Engl J Med 310: 559-562
59. Simioni P, Prandoni P, Zanon E, Saracino MA, et al (1996) Deep venous thrombosis and lupus anticoagulant. Thromb Haemost 76: 187-189
60. Simioni P, Tormene D, Manfrin D, et al (1998) Prothrombin antigen levels in symptomatic and asymptomatic carriers of the 20210A prothrombin variant. Br J Haematol 103: 1045-1050
61. Souto JC, Coll I, Llobet D, del Rio E, et al (1998) The prothrombin 20210A allele is the most present genetic risk factor for venous thromboembolism in the Spanish popu-lation. Thromb Haemost 80: 366-369
62. Svensson PJ, Dahlbäck B (1994) Resistance to activated protein C as a basis for ve-nous thrombosis. N Engl J Med 330: 517-522
63. Tans G, Curvers J, Middeldorp S, et al (2000) A randomized cross-over study on the effects of levonorgestrel- and desogestrel-containing oral contraceptives on the antico-agulant pathways. Thromb Haemost 84: 15-21
64. Thaler E, Lechner K (1981) Antithrombin III deficiency and thromboembolism. Clin Haematol 10: 369-390
65. Tosetto A, Castaman G, Cappelleri A, Rodeghiero F (2000) The VITA project: heritability of resistance of activated protein C. Thromb Haemost 84: 811-814
66. Turxtra F, Karemaker R, Kuijer PM, Prins MH, et al (1999) Is the prevalence of the factor V Leiden mutation in patients with pulmonary embolism and deep vein thrombo-sis really different? Thromb Haemost 81: 345-348
67. Van den Belt AGM, Sanson BJ, Simioni P, et al (1997) Recurrence of venous thromboembolism in patients with familial thrombophilia. Arch Intern Med 157: 2227-2232
68. Van Hylckama VA, van der Linden IK Bertina RM, Rosendaal FR (2000) High levels of factor IX increase the risk of venous thrombosis. Blood 95: 3678-3682
69. van Tilburg HN, Rosendaal FR, Bertina RM (2000) Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood 95: 2855-2859
70. Vandenbroucke JP, Bertina RM, Homes ZR, Spaargaren C, et al (1998) Factor V Leiden and fatal pulmonary embolism. Thromb Haemost 79: 511-516
71. Wahl DG, Guillemin F, de Maistre E, Perret C, et al (1997) Risk for venous thrombosis related to antiphospholipid-antibodies in systemic lupus erythematosus - a meta-analysis. Lupus 6: 467-473
72. Wahl DG, Guillemin F, de Maistre E, Perret-Guillaume C, et al (1998) Meta-analysis of the risk of venous thrombosis in individuals with antiphospholipid antibodies without underlying autoimmune disease or previous thrombosis. Lupus 7: 15-22
73. Wells PS, Anderson DR, Ginsberg J (2000) Assessment of deep vein thrombosis or pulmonary embolism by combined use of clinical model and non-invasive diagnostic tests. Sem Thromb Hemost 26: 643-641
74. Williamson D, Brown K, Luddington R, Baglin C, et al (1998) Factor V Cambridge: a new mutation (ARG306®Thr) associated with resistance to activated protein C Blood 91: 1140-1144
zum Anfang
zurück
